Efficient Autoencoder Pipeline for Discovering High Entropy Alloys with Molecular Dynamics Data
{kind=link}
Abstract
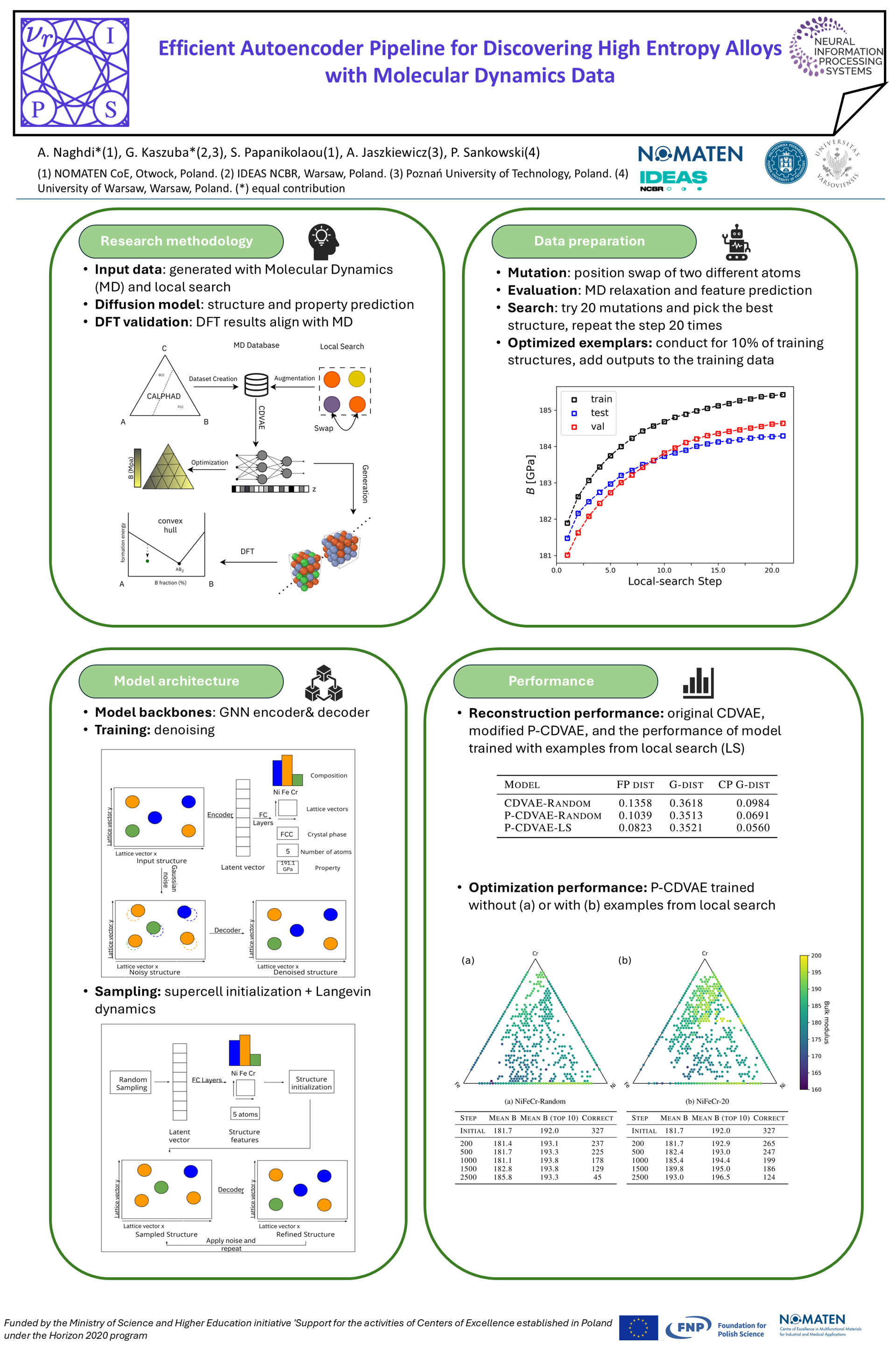
In this work, we utilize computationally efficient Molecular Dynamics (MD)simulations to create a machine learning pipeline for discovery of crystalline multi-component alloys. We employ high-quality interatomic potentials to create adataset of NiFeCr structures and apply Crystal Diffusion Variational Autoencoder(CDVAE) to maximize their mechanical properties, i.e. bulk modulus. As part ofthe experiment, we utilize local search coupled with classical interatomic potentialsto explore the local structure space and show that utilization of this proceduregreatly improves optimization capability of the neural model. We also expand themodel with an extra submodule, which attains 42% improvement on modeling thecrystalline phase of the structures. Ultimately, we verify the global stability of thecreated structures with quantum mechanical calculation methods.