DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
{kind=link}
Abstract
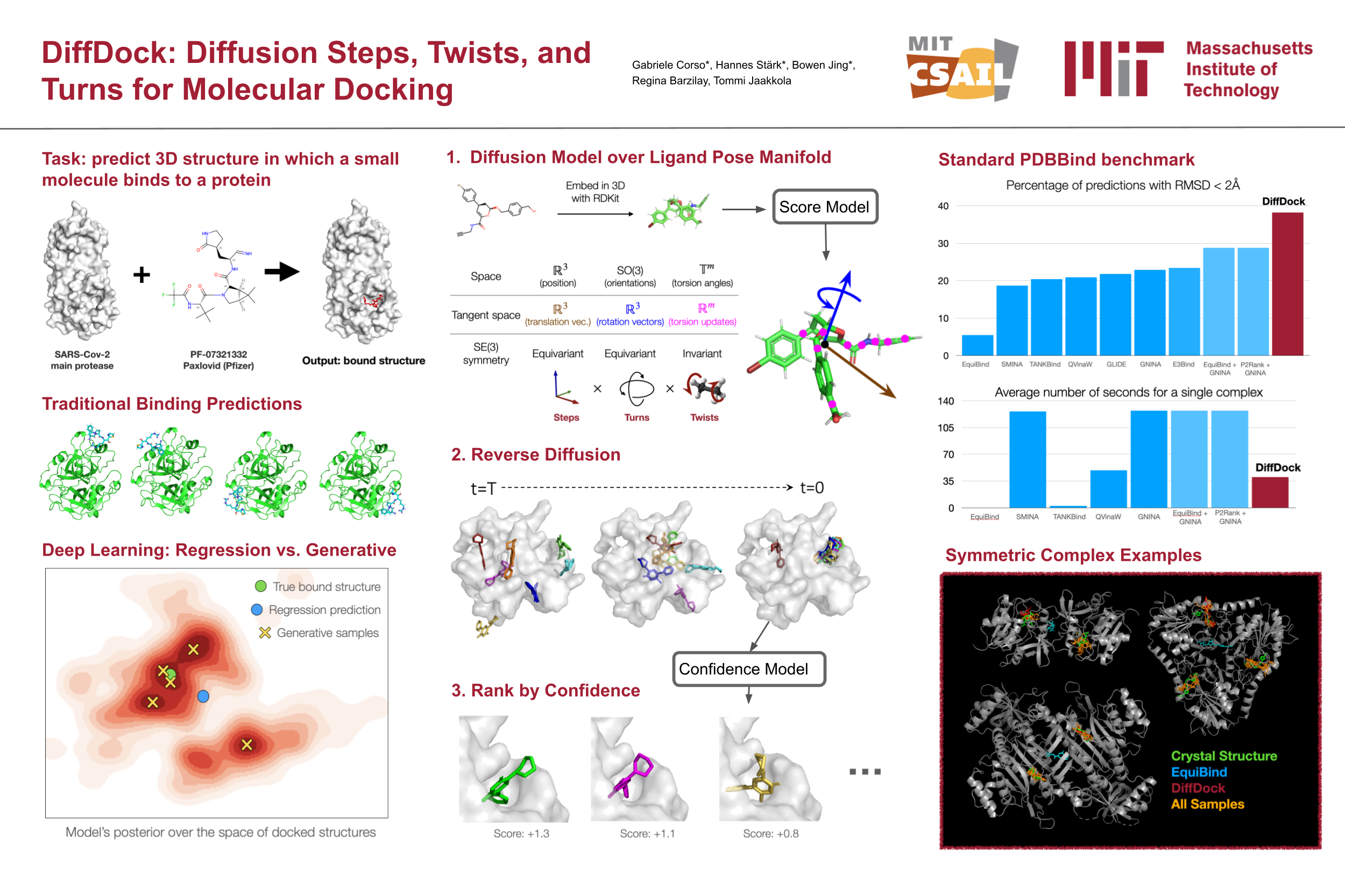
Predicting the binding structure of a small molecule ligand to a protein---a task known as molecular docking---is critical to drug design. Recent deep learning methods that treat docking as a regression problem have decreased runtime compared to traditional search-based methods but have yet to offer substantial improvements in accuracy. We instead frame molecular docking as a generative modeling problem and develop DiffDock, a diffusion generative model over the non-Euclidean manifold of ligand poses. To do so, we map this manifold to the product space of the degrees of freedom (translational, rotational, and torsional) involved in docking and develop an efficient diffusion process on this space. Empirically, DiffDock obtains a 38% top-1 success rate (RMSD<2Å) on PDBBind, significantly outperforming the previous state-of-the-art of traditional docking (23%) and deep learning (20%) methods. Moreover, DiffDock has fast inference times and provides confidence estimates with high selective accuracy.