Transfer Learning Lithium and Electrolyte Potential Energy Surfaces from Pure and Hybrid DFT
{kind=link}
Abstract
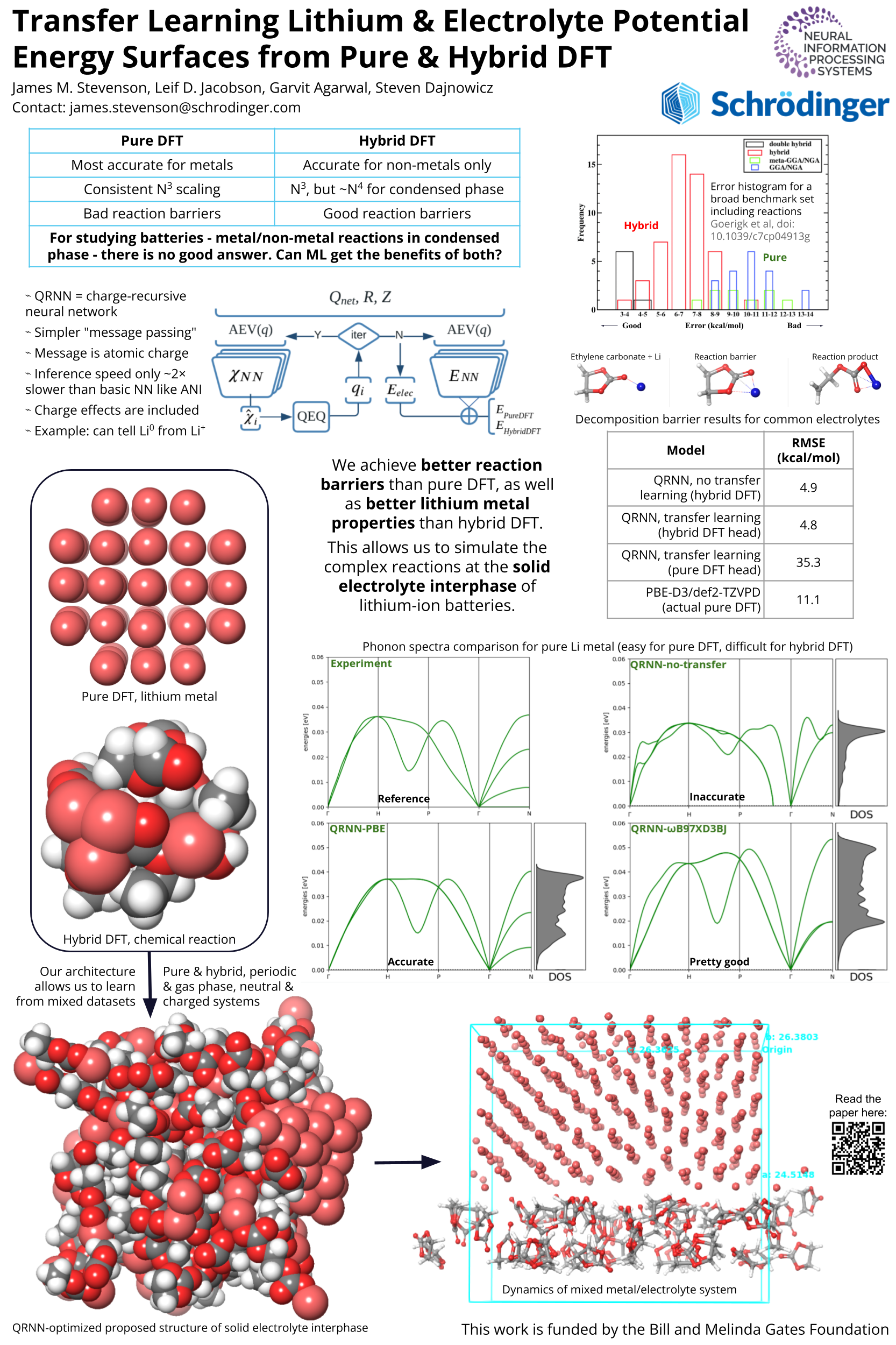
One of the most important problems in rational design of batteries is predicting the properties of the Solid Electrolyte Interphase, which (for a metallic anode) is the part of the battery where metallic and non-metallic components come into contact. However, there is a fundamental problem with predicting the properties of such a mixed material: the two components are best simulated with incompatible levels of density functional theory. Pure functionals perform well for metallic properties, while hybrid or long-range-corrected density functionals perform better for molecular properties and reaction barriers. We demonstrate a simple method to obviate this conflict by training a machine learning potential energy surface using both levels of theory via transfer learning. We further show that the resulting model is more accurate than models trained individually to these levels of theory, allowing more accurate property prediction and potentially faster materials discovery.