Protein-Protein Docking with Iterative Transformer
{kind=link}
Abstract
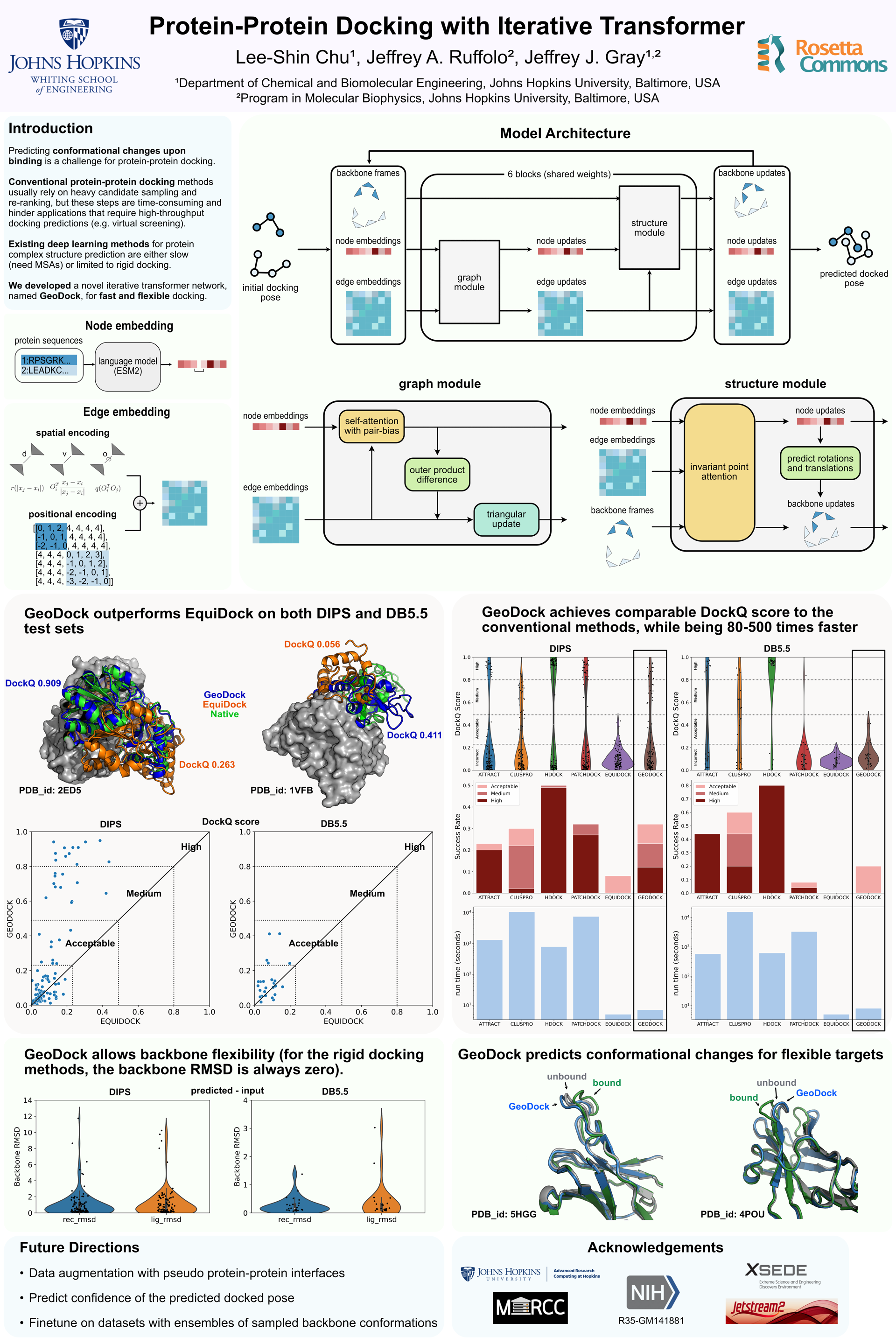
Conventional protein-protein docking algorithms usually rely on heavy candidate sampling and re-ranking, but these steps are time-consuming and hinder applications that require high-throughput complex structure prediction, e.g., structure-based virtual screening. Existing deep learning methods for protein-protein docking, despite being much faster, suffer from low docking success rates. In addition, they simplify the problem to assume no conformational changes within any protein upon binding (rigid docking). This assumption precludes applications when binding-induced conformational changes play a role, such as allosteric inhibition or docking from uncertain unbound model structures. To address the limitations, we designed a novel iterative transformer network that predicts the 3D transformation from a randomized initial docking pose to a refined docked pose. Our method, GeoDock, is flexible at the protein residue level, allowing the prediction of rigid-body movement as well as conformational changes upon binding. For two benchmark sets of rigid docking targets, GeoDock successfully docks 32% and 20% of the protein pairs, outperforming the baseline deep learning method EQUIDOCK (8% and 0% success rates). Additionally, GeoDock achieves comparable docking success rates to the conventional docking algorithms while being 80-500 times faster. Although binding-induced conformational changes are still a challenge owing to limited training and evaluation data, our architecture sets up the foundation to capture flexibility going ahead.