Using domain-domain interactions to probe the limitations of MSA pairing strategies
{kind=link}
Abstract
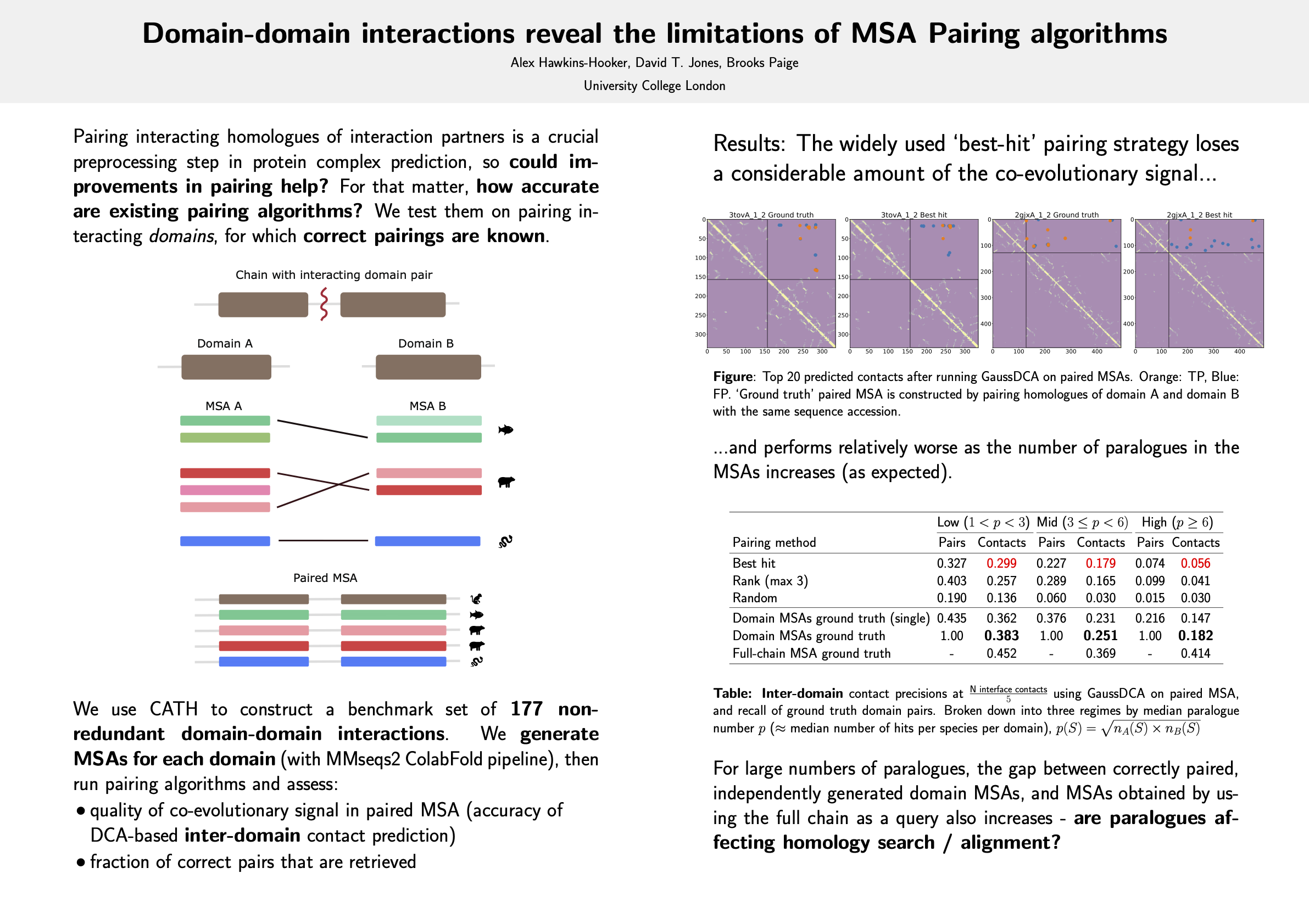
State-of-the-art methods for the prediction of the structures of interacting protein complexes rely on the construction of paired multiple sequence alignments, whose rows contain concatenated pairs of homologues of each of the interacting chains. Despite the inherent difficulty of accurately pairing interacting homologues of each chain, most existing methods use simple heuristic strategies for this purpose. The accuracy of these heuristic strategies and the consequences of their widespread usage remain poorly understood, due in large part to the paucity of ground truth data on correct pairings. To remedy this situation we propose a novel benchmark setting for interaction partner pairing algorithms, based on domain-domain interactions within single protein chains. The co-existence of pairs of domains within single chains means that ground-truth pairs of homologues are known a priori, allowing both the accuracy of pairing strategies and the influence of inaccurate pairings on downstream inferences to be quantified directly. We provide evidence that the widely used best-hit pairing strategy leads in many cases to very noisy paired MSAs, from which inferences of 3D structure can be significantly less accurate than those made using the correctly paired MSAs. We conclude that further improvements in pairing strategies promise significant benefits for structure predictors capable of exploiting co-evolutionary signal.